Enzymes (pronounced /ˈɛnzaɪmz/) are proteins that catalyze (i.e., increase the rates of) chemical reactions.[1][2] In enzymatic reactions, the molecules at the beginning of the process, called substrates, are converted into different molecules, called products. Almost all chemical reactions in a biological cell need enzymes in order to occur at rates sufficient for life. Since enzymes are selective for their substrates and speed up only a few reactions from among many possibilities, the set of enzymes made in a cell determines which metabolic pathways occur in that cell.
Like all catalysts, enzymes work by lowering the activation energy (Ea‡) for a reaction, thus dramatically increasing the rate of the reaction. As a result, products are formed faster and reactions reach their equilibrium state more rapidly. Most enzyme reaction rates are millions of times faster than those of comparable un-catalyzed reactions. As with all catalysts, enzymes are not consumed by the reactions they catalyze, nor do they alter the equilibrium of these reactions. However, enzymes do differ from most other catalysts in that they are highly specific for their substrates. Enzymes are known to catalyze about 4,000 biochemical reactions.[3] A few RNA molecules called ribozymes also catalyze reactions, with an important example being some parts of theribosome.[4][5] Synthetic molecules called artificial enzymes also display enzyme-like catalysis.[6]
Enzyme activity can be affected by other molecules. Inhibitors are molecules that decrease enzyme activity; activators are molecules that increase activity. Many drugs and poisons are enzyme inhibitors. Activity is also affected by temperature, chemical environment (e.g., pH), and the concentration of substrate. Some enzymes are used commercially, for example, in the synthesis of antibiotics. In addition, some household products use enzymes to speed up biochemical reactions (e.g., enzymes in biological washing powders break down protein or fat stains on clothes; enzymes in meat tenderizers break down proteins into smaller molecules, making the meat easier to chew).
Etymology and history
As early as the late 17th and early 18th centuries, the digestion of meat by stomach secretions[7] and the conversion of starch to sugars by plant extracts and saliva were known. However, the mechanism by which this occurred had not been identified.[8]
In the 19th century, when studying the fermentation of sugar to alcohol by yeast, Louis Pasteur came to the conclusion that this fermentation was catalyzed by a vital force contained within the yeast cells called "ferments", which were thought to function only within living organisms. He wrote that "alcoholic fermentation is an act correlated with the life and organization of the yeast cells, not with the death or putrefaction of the cells."[9]
In 1877, German physiologist Wilhelm Kühne (1837–1900) first used the term enzyme, which comes from Greek ενζυμον, "in leaven", to describe this process.[10] The word enzymewas used later to refer to nonliving substances such as pepsin, and the word ferment was used to refer to chemical activity produced by living organisms.
In 1897, Eduard Buchner submitted his first paper on the ability of yeast extracts that lacked any living yeast cells to ferment sugar. In a series of experiments at the University of Berlin, he found that the sugar was fermented even when there were no living yeast cells in the mixture.[11] He named the enzyme that brought about the fermentation of sucrose "zymase".[12] In 1907, he received the Nobel Prize in Chemistry "for his biochemical research and his discovery of cell-free fermentation". Following Buchner's example, enzymes are usually named according to the reaction they carry out. Typically, to generate the name of an enzyme, the suffix -ase is added to the name of its substrate (e.g., lactase is the enzyme that cleaves lactose) or the type of reaction (e.g., DNA polymerase forms DNA polymers).[13]
Having shown that enzymes could function outside a living cell, the next step was to determine their biochemical nature. Many early workers noted that enzymatic activity was associated with proteins, but several scientists (such as Nobel laureate Richard Willstätter) argued that proteins were merely carriers for the true enzymes and that proteins per sewere incapable of catalysis.[citation needed] However, in 1926, James B. Sumner showed that the enzyme urease was a pure protein and crystallized it; Sumner did likewise for the enzyme catalase in 1937. The conclusion that pure proteins can be enzymes was definitively proved by Northrop and Stanley, who worked on the digestive enzymes pepsin (1930), trypsin and chymotrypsin. These three scientists were awarded the 1946 Nobel Prize in Chemistry.[14]
This discovery that enzymes could be crystallized eventually allowed their structures to be solved by x-ray crystallography. This was first done for lysozyme, an enzyme found in tears, saliva and egg whites that digests the coating of some bacteria; the structure was solved by a group led by David Chilton Phillips and published in 1965.[15] This high-resolution structure of lysozyme marked the beginning of the field ofstructural biology and the effort to understand how enzymes work at an atomic level of detail.
Structures and mechanisms
Enzymes are generally globular proteins and range from just 62 amino acid residues in size, for the monomer of 4-oxalocrotonate tautomerase,[16] to over 2,500 residues in the animal fatty acid synthase.[17] A small number of RNA-based biological catalysts exist, with the most common being the ribosome; these are referred to as either RNA-enzymes or ribozymes. The activities of enzymes are determined by their three-dimensional structure.[18] However, although structure does determine function, predicting a novel enzyme's activity just from its structure is a very difficult problem that has not yet been solved.[19]
Most enzymes are much larger than the substrates they act on, and only a small portion of the enzyme (around 3–4 amino acids) is directly involved in catalysis.[20] The region that contains these catalytic residues, binds the substrate, and then carries out the reaction is known as the active site. Enzymes can also contain sites that bind cofactors, which are needed for catalysis. Some enzymes also have binding sites for small molecules, which are often direct or indirect products or substrates of the reaction catalyzed. This binding can serve to increase or decrease the enzyme's activity, providing a means forfeedback regulation.
Like all proteins, enzymes are long, linear chains of amino acids that fold to produce a three-dimensional product. Each unique amino acid sequence produces a specific structure, which has unique properties. Individual protein chains may sometimes group together to form a protein complex. Most enzymes can be denatured—that is, unfolded and inactivated—by heating or chemical denaturants, which disrupt the three-dimensional structure of the protein. Depending on the enzyme, denaturation may be reversible or irreversible.
Structures of enzymes in complex with substrates or substrate analogs during a reaction may be obtained using Time resolved crystallography methods.
Specificity
Enzymes are usually very specific as to which reactions they catalyze and the substrates that are involved in these reactions. Complementary shape, charge and hydrophilic/hydrophobic characteristics of enzymes and substrates are responsible for this specificity. Enzymes can also show impressive levels of stereospecificity, regioselectivity and chemoselectivity.[21]
Some of the enzymes showing the highest specificity and accuracy are involved in the copying and expression of the genome. These enzymes have "proof-reading" mechanisms. Here, an enzyme such as DNA polymerase catalyzes a reaction in a first step and then checks that the product is correct in a second step.[22] This two-step process results in average error rates of less than 1 error in 100 million reactions in high-fidelity mammalian polymerases.[23] Similar proofreading mechanisms are also found in RNA polymerase,[24] aminoacyl tRNA synthetases[25] and ribosomes.[26]
Some enzymes that produce secondary metabolites are described as promiscuous, as they can act on a relatively broad range of different substrates. It has been suggested that this broad substrate specificity is important for the evolution of new biosynthetic pathways.[27]
"Lock and key" model
Enzymes are very specific, and it was suggested by the Nobel laureate organic chemist Emil Fischer in 1894 that this was because both the enzyme and the substrate possess specific complementary geometric shapes that fit exactly into one another.[28] This is often referred to as "the lock and key" model. However, while this model explains enzyme specificity, it fails to explain the stabilization of the transition state that enzymes achieve.

In 1958, Daniel Koshland suggested a modification to the lock and key model: since enzymes are rather flexible structures, the active site is continually reshaped by interactions with the substrate as the substrate interacts with the enzyme.[29] As a result, the substrate does not simply bind to a rigid active site; the amino acid side chains which make up the active site are molded into the precise positions that enable the enzyme to perform its catalytic function. In some cases, such as glycosidases, the substrate molecule also changes shape slightly as it enters the active site.[30] The active site continues to change until the substrate is completely bound, at which point the final shape and charge is determined.[31] Induced fit may enhance the fidelity of molecular recognition in the presence of competition and noise via the conformational proofreading mechanism .[32]
Mechanisms
Enzymes can act in several ways, all of which lower ΔG‡:[33]
- Lowering the activation energy by creating an environment in which the transition state is stabilized (e.g. straining the shape of a substrate—by binding the transition-state conformation of the substrate/product molecules, the enzyme distorts the bound substrate(s) into their transition state form, thereby reducing the amount of energy required to complete the transition).
- Lowering the energy of the transition state, but without distorting the substrate, by creating an environment with the opposite charge distribution to that of the transition state.
- Providing an alternative pathway. For example, temporarily reacting with the substrate to form an intermediate ES complex, which would be impossible in the absence of the enzyme.
- Reducing the reaction entropy change by bringing substrates together in the correct orientation to react. Considering ΔH‡ alone overlooks this effect.
- Increases in temperatures speed up reactions. Thus, temperature increases help the enzyme function and develop the end product even faster. However, if heated too much, the enzyme’s shape deteriorates and the enzyme becomes denatured. Some enzymes like thermolabile enzymes work best at low temperatures.
Interestingly, this entropic effect involves destabilization of the ground state,[34] and its contribution to catalysis is relatively small.[35]
Transition State Stabilization
The understanding of the origin of the reduction of ΔG‡ requires one to find out how the enzymes can stabilize its transition state more than the transition state of the uncatalyzed reaction. Apparently, the most effective way for reaching large stabilization is the use of electrostatic effects, in particular, when having a relatively fixed polar environment that is oriented toward the charge distribution of the transition state.[36]Such an environment does not exist in the uncatalyzed reaction in water.
Dynamics and function
See also: Protein dynamics
The internal dynamics of enzymes has been suggested to be linked with their mechanism of catalysis.[37][38][39] Internal dynamics are the movement of parts of the enzyme's structure, such as individual amino acid residues, a group of amino acids, or even an entire protein domain. These movements occur at various time-scales ranging from femtoseconds to seconds. Networks of protein residues throughout an enzyme's structure can contribute to catalysis through dynamic motions.[40][41][42][43] This is simply seen in the kinetic scheme of the combined process, enzymatic activity and dynamics; this scheme can have several independentMichaelis-Menten-like reaction pathways that are connected through fluctuation rates. [44][45][46]
Protein motions are vital to many enzymes, but whether small and fast vibrations, or larger and slower conformational movements are more important depends on the type of reaction involved. However, although these movements are important in binding and releasing substrates and products, it is not clear if protein movements help to accelerate the chemical steps in enzymatic reactions.[47] These new insights also have implications in understanding allosteric effects and developing new medicines.
Allosteric modulation

Main article: Allosteric regulation
Allosteric sites are sites on the enzyme that bind to molecules in the cellular environment. The sites form weak, noncovalent bonds with these molecules, causing a change in the conformation of the enzyme. This change in conformation translates to the active site, which then affects the reaction rate of the enzyme.[48] Allosteric interactions can both inhibit and activate enzymes and are a common way that enzymes are controlled in the body.[49]
Cofactors and coenzymes
Cofactors
Some enzymes do not need any additional components to show full activity. However, others require non-protein molecules called cofactors to be bound for activity.[50] Cofactors can be either inorganic (e.g., metal ions and iron-sulfur clusters) or organic compounds (e.g., flavin and heme). Organic cofactors can be eitherprosthetic groups, which are tightly bound to an enzyme, or coenzymes, which are released from the enzyme's active site during the reaction. Coenzymes include NADH, NADPH and adenosine triphosphate. These molecules transfer chemical groups between enzymes.[51]
An example of an enzyme that contains a cofactor is carbonic anhydrase, and is shown in the ribbon diagram above with a zinc cofactor bound as part of its active site.[52] These tightly bound molecules are usually found in the active site and are involved in catalysis. For example, flavin and heme cofactors are often involved in redox reactions.
Enzymes that require a cofactor but do not have one bound are called apoenzymes or apoproteins. An apoenzyme together with its cofactor(s) is called a holoenzyme (this is the active form). Most cofactors are not covalently attached to an enzyme, but are very tightly bound. However, organic prosthetic groups can be covalently bound (e.g., biotin in the enzyme pyruvate carboxylase). The term "holoenzyme" can also be applied to enzymes that contain multiple protein subunits, such as the DNA polymerases; here the holoenzyme is the complete complex containing all the subunits needed for activity.
Coenzymes

Coenzymes are small organic molecules that can be loosely or tightly bound to an enzyme. Tightly bound coenzymes can be called allosteric groups. Coenzymes transport chemical groups from one enzyme to another.[53] Some of these chemicals such as riboflavin, thiamine and folic acid are vitamins (compounds which cannot be synthesized by the body and must be acquired from the diet). The chemical groups carried include the hydride ion (H-) carried by NAD or NADP+, the phosphate group carried by adenosine triphosphate, the acetyl group carried by coenzyme A, formyl, methenyl or methyl groups carried by folic acid and the methyl group carried by S-adenosylmethionine.
Since coenzymes are chemically changed as a consequence of enzyme action, it is useful to consider coenzymes to be a special class of substrates, or second substrates, which are common to many different enzymes. For example, about 700 enzymes are known to use the coenzyme NADH.[54]
Coenzymes are usually continuously regenerated and their concentrations maintained at a steady level inside the cell: for example, NADPH is regenerated through the pentose phosphate pathway and S-adenosylmethionine by methionine adenosyltransferase. This continuous regeneration means that even small amounts of coenzymes are used very intensively. For example, the human body turns over its own weight in ATP each day
Thermodynamics
As all catalysts, enzymes do not alter the position of the chemical equilibrium of the reaction. Usually, in the presence of an enzyme, the reaction runs in the same direction as it would without the enzyme, just more quickly. However, in the absence of the enzyme, other possible uncatalyzed, "spontaneous" reactions might lead to different products, because in those conditions this different product is formed faster.
Furthermore, enzymes can couple two or more reactions, so that a thermodynamically favorable reaction can be used to "drive" a thermodynamically unfavorable one. For example, the hydrolysis of ATP is often used to drive other chemical reactions.[56]
Enzymes catalyze the forward and backward reactions equally. They do not alter the equilibrium itself, but only the speed at which it is reached. For example,carbonic anhydrase catalyzes its reaction in either direction depending on the concentration of its reactants.
 (in
(in 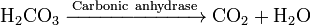 (in
(in Nevertheless, if the equilibrium is greatly displaced in one direction, that is, in a very exergonic reaction, the reaction is effectively irreversible. Under these conditions the enzyme will, in fact, only catalyze the reaction in the thermodynamically allowed direction.
No comments:
Post a Comment